The Bio Research Stack: Automating the Pipeline, Not the Science
How life sciences researchers and bioinformaticians use Claude Code skills for single-cell RNA-seq, Nextflow pipelines, scVI analysis, and instrument data workflows.

Research generates two kinds of work: the science — the experimental design, the interpretation, the insight — and the computational infrastructure that makes it possible. The second category is real engineering work, and it takes real time away from the first. Environment setup on a new cluster, QC parameter decisions on a new dataset, Nextflow configuration for a pipeline you run twice a year — none of it requires biological expertise, but all of it requires enough technical knowledge that it can’t be delegated without detailed documentation that rarely exists.
The Bio Research Stack is built for the computational layer: pipeline development, QC workflows, deep learning tools, and data standardisation. These are the tasks that are methodologically well-established, follow documented best practices, and benefit from consistent execution across datasets and projects. Claude handles this layer consistently — the same QC parameters, the same pipeline flags, the same data conversion conventions — without you having to re-specify the setup from scratch each session.
What the stack doesn’t touch is the science itself: the biological interpretation of a QC plot, the decision about which cell types to annotate, the experimental design choice that determines whether a dataset will answer the question you’re asking. Those decisions stay with the researcher. The stack’s job is to compress the time between data generation and the point where the scientific judgment calls actually begin.
The stack, workflow by workflow
Starting a project
Bio Research Start structures the beginning of a new research project — defining the scientific question, mapping the experimental approach, identifying the key computational dependencies, and the project framing that keeps a complex multi-step program on track from day one. Starting a new project in a new computational environment involves a predictable set of setup steps that take longer than they should: tool discovery, environment configuration, dependency resolution, and getting the first workflow running in a clean environment. The skill handles this setup layer systematically so the first pipeline run is a checkpoint, not a debugging session.
Use it at the start of a new project, when onboarding into a new computational environment you haven’t worked in before, or when setting up a fresh cluster allocation or cloud instance for a pipeline that needs to run in a clean environment.
Bio Research Scientific Problem Selection evaluates whether a research question is worth pursuing — novelty assessment against the literature, feasibility analysis given available data and compute, impact framing, and the structured problem selection process that separates tractable questions from ones that sound compelling but aren’t. Most scientists evaluate new project ideas informally — through conversation, intuition, and a rough mental model of feasibility. A structured framework surfaces the considerations that tend to get missed in that process: whether similar analyses have been published, whether the data quality is sufficient, whether the computational approach is matched to the question.
Use it when evaluating a new project direction before committing significant resources, when deciding between competing analytical approaches for the same dataset, or when a project has stalled and you need a structured framework for diagnosing whether it’s worth continuing.
npx skills add anthropics/knowledge-work-plugins --skill bio-researchSingle-cell RNA sequencing
Bio Research Single Cell RNA QC runs quality control on scRNA-seq data — cell filtering based on per-cell quality metrics, doublet detection, ambient RNA removal, mitochondrial fraction assessment using MAD-based adaptive thresholds, and comprehensive QC visualisations following scverse best practices. The scverse ecosystem has standardised QC methodology, but implementing it correctly across different datasets still requires knowing which parameters to set and why, and producing the visualisations that make QC decisions defensible. MAD-based filtering adjusts thresholds to the distribution of each dataset rather than applying fixed cutoffs that may be appropriate for one sample type and wrong for another.
Accepts .h5ad or .h5 files. The output is clean, analysis-ready data with a documented QC report — violin plots, scatter plots, and distribution histograms that document the filtering decisions and make them reproducible.
Use it as the first step after receiving new sequencing data before any downstream analysis, when applying consistent QC standards across a multi-sample dataset where thresholds need to adapt to each sample’s distribution, or when following scverse best practices is required for publication or reproducibility.
Bio Research scVI Tools applies variational autoencoder-based analysis to single-cell data — latent space modelling with scVI for dimensionality reduction and batch correction, semi-supervised cell type classification with scANVI, ATAC-seq analysis with PeakVI, CITE-seq multi-modal integration with totalVI, and multiome analysis with MultiVI. The scVI model family handles the analytical challenges where classical approaches break down: large-scale batch correction across datasets from different protocols, multi-modal integration where the feature spaces are heterogeneous, and reference mapping when you have a well-characterised atlas to map new data onto.
These models are more complex to configure and interpret correctly than PCA-based workflows — the training parameters, the number of latent dimensions, and the covariate specification all affect the quality of the learned representation. The skill covers model configuration, training, latent space interpretation, and the downstream analyses that build on the learned representations.
Use it for batch correction across datasets from different protocols or batches, for multi-modal data integration in CITE-seq or multiome experiments, or for reference mapping when a well-characterised reference atlas exists for your cell type of interest.
npx skills add anthropics/knowledge-work-plugins --skill bio-researchPipeline development
Bio Research Nextflow Development develops and maintains Nextflow bioinformatics pipelines — workflow structure, process definitions, channel logic, executor configuration for HPC and cloud environments, and nf-core pipeline integration. Running nf-core pipelines correctly requires knowing the right pipeline version, the correct parameter flags for your data type, how to handle input samplesheets, and how to configure execution for your compute environment. Getting it wrong wastes compute time and produces results you can’t trust — and the error often isn’t obvious until you compare outputs against a known-good reference.
Can run established nf-core pipelines — rnaseq, sarek, atacseq — on local FASTQs or public datasets from GEO and SRA. Covers pipeline configuration for your compute environment, samplesheet generation, the key parameters that affect output quality, and the post-run checks that confirm the pipeline completed correctly before you commit to downstream analysis.
Use it when setting up a bulk sequencing pipeline for the first time on a new cluster or cloud environment, when running a nf-core pipeline you use infrequently enough that the parameter choices aren’t memorised, or when debugging a pipeline run that completed with errors or produced unexpected output.
npx skills add anthropics/knowledge-work-plugins --skill bio-researchInstrument data standardisation
Bio Research Instrument Data to Allotrope converts laboratory instrument output files — PDF, CSV, Excel, TXT — to Allotrope Simple Model (ASM) JSON or flattened 2D CSV. Laboratory instrument outputs are a Tower of Babel: each instrument produces its own proprietary format that requires custom parsing before it can be used in downstream systems. The Allotrope format is the emerging standard for instrument data interoperability in regulated environments, and conversion is the prerequisite for LIMS integration or any analysis pipeline that ingests data from multiple instrument types.
The skill handles parsing the instrument-specific format, mapping fields to the Allotrope schema, handling missing or ambiguous fields, and validating the output against the target schema — the steps that require knowing the format specification rather than biological domain knowledge.
Use it when standardising instrument data for a LIMS system, when building a data pipeline that ingests data from multiple instrument types with incompatible native formats, or when a downstream analysis workflow requires a consistent data format regardless of which instrument generated the data.
npx skills add anthropics/knowledge-work-plugins --skill bio-researchHow the stack works together
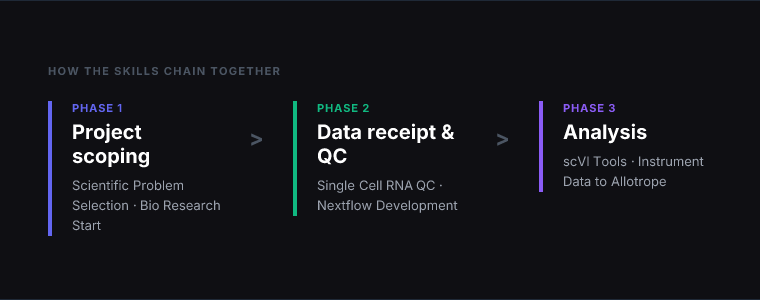
The skills map to a typical single-cell RNA-seq project from data receipt to analysis. Here’s how they chain across the full workflow:
Project scoping: Use Scientific Problem Selection before committing to a new analytical direction — particularly useful when deciding between competing approaches or evaluating whether the available data is sufficient to answer the question.
Environment setup: Use Bio Research Start when setting up the computational environment on a new cluster, cloud instance, or collaborator’s system. This runs once per environment, not once per project.
Bulk sequencing: Use Nextflow Development for any bulk sequencing data — RNA-seq, ATAC-seq, variant calling — that feeds into the single-cell analysis or stands alone as a project deliverable. Run this before the single-cell QC if the data hasn’t been processed from raw reads.
Single-cell data receipt: Use Single Cell RNA QC as the first step after receiving new sequencing data. Run the QC, review the visualisations, make the filtering decisions, and document the thresholds before any downstream analysis begins.
Dimensionality reduction and integration: Use scVI Tools after QC if the dataset spans multiple batches, protocols, or modalities. For a single-batch, single-modality dataset, classical dimensionality reduction may be sufficient — the scVI skill is most valuable where batch effects are large enough to confound downstream analysis.
Instrument data: Use Instrument Data to Allotrope when lab instrument outputs need to be standardised before entering the analysis pipeline. This is a prerequisite for LIMS integration and for any workflow where data from multiple instrument types needs to be compared.
The install command is the same for all six skills — --skill bio-research from the anthropics/knowledge-work-plugins repo. Each skill operates independently and triggers on context. You don’t need to use all of them — most projects use three or four depending on the data type and the computational environment.
Install the full stack
Browse all research skills → /audiences/researchers
📬 Weekly digest
Get the best new skills every Tuesday
3–5 hand-picked skills. Free forever.
More guides
April 4, 2026 · 8 min read
The CEO of Y Combinator Ships 10,000 Lines of Code a Day. Here's Exactly How.
Garry Tan runs one of the most demanding jobs in tech. He's also shipping more code than ever. gstack — his open-source Claude Code system — is how. Here's what it is and why it works.
March 25, 2026 · 7 min read
How to Create a Claude Skill (Step-by-Step Guide)
Learn how to build, test, and share your own Claude Code skills. A complete walkthrough — from blank file to installed skill — with real examples and best practices.
March 24, 2026 · 8 min read
Awesome Claude Skills: The Complete Searchable List (2026)
Every major Claude Code skills list and awesome-claude-code repository in one place — with install commands, categories, and a searchable directory for all 370+ skills.